
Abstract
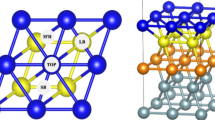
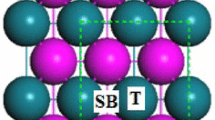
Periodic density functional theory (DFT) calculations were performed to investigate the adsorption of H2O on U(001) surface. The metallic nature of uranium atom and different adsorption sites of U(001) surface play key roles in the H2O molecular dissociate reaction. The long-bridge site is the most favorable site of H2O-U(001) adsorption configuration. The triangle-center site of the H atom is the most favorable site of HOH-U(001) adsorption configuration. The interaction between H2O and U surface is more evident on the first layer than that on any other two sub-layers. The dissociation energy of one hydrogen atom from H2O is −1.994 to −2.215 eV on U(001) surface, while the dissociating energy decreases to −3.351 to −3.394 eV with two hydrogen atoms dissociating from H2O. These phenomena also indicate that the Oads can promote the dehydrogenation of H2O. A significant charge transfer from the first layer of the uranium surface to the H and O atoms is also found to occur, making the bonding partly ionic.






Similar content being viewed by others

References
Soderlind P, Johansson B (1995) Nature 374(6522):524525
Lyon SB (2010) Shreirs. Corrosion 21812191
Mattox D, Bland R (1967) J Nucl Mater 21(3):349352
Long Z, Liu K, Bai B, Yan D (2010) J Alloys Compd 491(1):252257
Winer K, Colmenares CA, Smith RL (1987) Surf Sci 183(1):6799
Ritchie AG (1984) J Nucl Mater 120(2):143153
Allen GC, Tucker PM, Lewis RA (1984) J Chem Soc Faraday Trans 8(8):9911000
Mcgillivray GW (1994) J Nucl Mater 208(1):8197
Bloch J, Mintz MH (1981) J Less Common Met 81(2):301320
McLean W, Colmenares C, Smith R, Somorjai G (1982) Phys Rev B 25(1):8
Wang X, Fu Y, Xie R (1998) J Nucl Tech 21(4):233237
Li G, Luo W, Chen H (2010) Chem Res Appl 22(10):12831289
Xiulin Z, Shanqisong H, Xuehai J (2013) J Radional Nucl Chem 298:481484
Nie J, Xiao H, Zu XT, Gao F. J Phys Condens Matter 20(44): 445001
Ray A, Dholabhai P (2007) J Alloys Compd 356:31
BiTao X, DaQiao M, WeiDong X, ZhengHe Z, Gang J, HongYan W (2003) Acta Phys Sin 52(7):16171623
Li G, Luo W, Chen H (2011) Acta Phys -Chim Sin 27(10):23192325
Huda MN, Ray AK (2005) Int J Quantum Chem 102:098105
Taylor CD (2008) Phys Rev B 77:094119
Kozimor SA, Yang P, Batista ER, Boland KS, Burns CJ, Clark DL, Conradson SD, Martin RL, Wilkerson MP, Wolfsberg LE (2009) J Am Chem Soc 131(34):1212512136
Su QL, Deng HQ, Ao BY, Xiao SF, Li XF, Chen PH, Hu WY (2014) J Appl Phys 115:123
Zhao JY, Zhao FQ, Gao HX (2013) J Mol Model 4:19
Balasubramanian K, Siekhaus WJ (2003) J Chem Phys 119(12):58895900
Allen GC, Crofts JA, Curtis MT (1974) J Chem Soc Dalton Trans 12(12):12961301
Nie JL, Xiao HY, Gao F (2009) J Alloys Compd 476(1):675682
Suvorov AL (1977) Sov At Energy 42(4):312317
Liu R (2013) Comput Theor Chem 1019:141145
Acknowledgments
This work was financially supported by the National Science Foundation of China (Grant No. 21101070) and the Funding from the Science and Technology on Combustion and Explosion Laboratory.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Huang, S., Zeng, XL., Zhao, FQ. et al. Density functional study of H2O molecule adsorption on α-U(001) surface. J Mol Model 22, 88 (2016). https://doi.org/10.1007/s00894-016-2956-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-016-2956-6