
Abstract
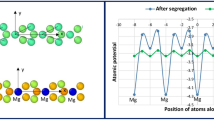
The atomic structures and energetics of clean and Y-doped general grain boundary (GB) ∑31/(0001) models in α-Al2O3 are studied by a series of high precision ab initio calculations. A large supercell with 700 atoms and periodic boundary conditions is adopted for undoped and Y-doped GB with different substitution sites and concentrations. It is shown that Y atoms preferably segregate to the central column of the 7-member Al ring. This is explained as more favorable bond formation for Y in this position and lower GB energy. The calculated GB formation energy for the clean and Y-doped cases is respectively 3.99 and 3.67 J/m2. On the average, the GB region in ∑31 has a slightly lower charge density than the bulk crystalline region. In addtition, the GB induces a long ranged asymmetric electrostatic potential distribution on each side of the grain boundary.
Similar content being viewed by others


References
Cannon R M, Rhodes W H, Heuer A H. Plastic-deformation of fine-grained alumina (Al2O3): Interface-controlled diffusional creep. J Am Ceram Soc, 1980, 63: 46–53
Hoche T, Kenway P R, Kleebe H J, et al. The structure of special grain boundaries in α-Al2O3. J Phys Chem Solids, 1994, 55(10): 1067–1082
Cho J, Harman M P, Chan H M, et al. Effect of yttrium and lanthanum on the tensile creep behavior of aluminum oxide. J Am Ceram Soc, 1997, 80(4): 1013–1017
Yoshida H, Ikuhara Y, Sakuma T. Grain boundary electronic structure related to the high-temperature creep resistance in polycrystalline Al2O3. Acta Mater, 2002, 50: 2955–2966
Lartigue-Korinek S, Priester L. Comment on influence of yttrium doping on grain misorientation in aluminium oxide. J Am Ceram Soc, 2000, 83: 1323
Atlay A, Gulgun M A. Microstructural evolution of calcium-doped alumina. J Am Ceram Soc, 2003, 86(4): 623–629
Matsunaga K, Nishimura H, Hanyu S, et al. HRTEM study on grain boundary atomic structures related to the sliding behavior in alumina bicrystals. Appl Surf Sci, 2005, 241: 75–79
Hanyu S, Nishimura H, Matsunaga K, et al. Stress-induced facet coarsening in a ∑7{4510} symmetrical tilt grain boundary in an alumina bicrystal. J Mater Sci, 2005, 40: 3137–3142
Matsunaga K, Nishimura H, Muto H, et al. Direct measurements of grain boundary sliding in yttrium-doped alumina bicrystals. Appl Phys Lett, 2003, 82: 1179–1181
Nishimura H, Matsunaga K, Saito T, et al. Atomic structures and energies of ∑7 symmetrical tilt grain boundaries in alumina bicrystals. J Am Ceram Soc, 2003, 86(4): 574–580
Marinopoulos A G, Nufer S, Elsässer C. Interfacial structures and energetics of basal twins in α-Al2O3: First-principles density-functional and empirical calculations. Phys Rev B, 2001, 63: 165112–165121
Fabris S, Elsässer C. First-principles analysis of cation segregation at grain boundaries in α-Al2O3. Acta Mater, 2003, 51: 71–86
Chen J, Xu Y N, Rulis P, et al. Ab initio theoretical tensile test on Y-doped ∑ = 3 grain boundary in α-Al2O3. Acta Mater, 2005, 53: 403–410
Chen J, Ouyang L, Ching W Y. Molecular dynamics simulation of Y-doped ∑37 grain boundary in alumina. Acta Mater, 2005, 53: 4111–4120
Northrup J E, Neugebauer J, Romano L T. Inversion domain and stacking mismatch boundaries in GaN. Phys Rev Lett, 1996, 77: 103–106
Kohyama M. Ab initio study of the tensile strength and fracture of coincidence tilt boundaries in cubic polar interfaces of the {122} ∑= 9 boundary SiC. Phys Rev B, 2002, 65: 184107–184118
Zhang Y, Lu G H, Deng S, et al. Weakening of an aluminum grain boundary induced by sulfur segregation: A first-principles computational tensile test. Phys Rev B, 2007, 75: 174101
Lu G H, Zhang Y, Deng S, et al. Origin of intergranular embrittlement of Al alloys induced by Na and Ca segregation: Grain boundary weakening. Phys Rev B, 2006, 73: 224115
Yamaguchi M, Shiga M, Kaburaki H. Grain boundary decohesion by impurity segregation in a nickel-sulfur system. Science, 2005, 307: 393–397
Elsässer C, Elaasser T. Codoping and grain-boundary cosegregation of substitutional cations in Al2O3: A density-functional-theory study. J Am Ceram Soc, 2005, 88(1): 1–14
Buban J, Matsunaga K, Chen J, et al. Grain boundary strengthening in alumina by rare earth impurities. Science, 2006, 311: 212–214
Catlow C R A, James R. Defect energetics in α-Al2O3 and rutile TiO2. Phys Rev B, 1982, 25(2): 1006–1026
Gale D. “GULP”, a general utility lattice program. http://www.ch.ic.ac.uk/gale/Research/gulp.html, 2002-08
Brandon D G, Ralph B, Rangantahn S, et al. A field ion microscope study of atomic configuration at grain boundaries. Acta Metal, 1964, 12: 813–821
Grimmer H, Bonnet R, Latigue S, et al. Theoretical and experimental description of grain boundaries in rhombohedral α-Al2O3. Phil Mag A, 1990, 61: 493–509
Kresse G, Furthmuller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plain wave basis set. Comput Mat Sci, 1996, 6: 15–50
Ching W Y. Electronic structure and bonding of all crystalline phase in the silica-yttria-silicon nitride phase equilibrium diagram. J Am Ceram Soc, 2004, 87(11): 1996–2013
Kingery W D. Plausible concepts necessary and sufficient for interpretation of ceramic grain boundary phenomena (I): Grain boundary characteristics, structure and electrostatic potential. J Am Ceram Soc, 1974, 57(1): 1–8
Ikeda J A S, Chiang Y M. Space charge segregation at grain boundaries in titanium dioxide (I): Relationship between lattice defect chemistry and space charge potential. J Am Ceram Soc, 1993, 76(10): 2437–2446
Ikeda J A S, Chiang Y M, Garratt-Reed A J, et al. Space charge segregation at grain boundaries in titanium dioxide (II): Model experiments. J Am Ceram Soc, 1993, 76(10): 2447–2459
Kliewer K L, Kohler J S. Space charge in ionic crystals (I): General approach with application to NaCl. Phys Rev, 1965, 140: A1226–A1240
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the National Natural Science Foundation of China (Grant Nos. 10744002 and 10774017)
Rights and permissions
About this article
Cite this article
Chen, J., Xu, Y., Chen, D. et al. Ab initio study of a Y-doped ∑31 grain boundary in alumina. Sci. China Ser. G-Phys. Mech. Astron. 51, 1607–1615 (2008). https://doi.org/10.1007/s11433-008-0154-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11433-008-0154-y