
Abstract
The use of the traditional xenograft subcutaneous tumor model has been contested because of its limitations, such as a slow tumorigenesis, inconsistent chemotherapeutic results, etc. In light of these challenges, we aim to revamp the traditional model by employing an electrospun scaffold composed of polydioxanone, gelatin and elastin to boost the tumorigenesis. The scaffold featured a highly porous microstructure and successfully supported the growth of tumor cells in vitro without provoking apoptosis. In vivo studies showed that in the scaffold model the tumor volume increased by 43.27% and the weight by 75.58%, respectively, within a 12-week period. In addition, the scaffold model saw an increase of CD24+ and CD44+ cells in the tumor mass by 42% and 313%, respectively. The scaffolding materials did not lead to phenotypic changes during the tumorigenesis. Thereafter, in the scaffold model, we found that the chemotherapeutic regimen of docetaxel, cisplatin and fluorouracil unleashed a stronger capability than the regimen comprising cisplatin and fluorouracil to deplete the CD44+ subpopulation. This discovery sheds mechanistic lights on the role of docetaxel for its future chemotherapeutic applications. This revamped model affords cancer scientists a convenient and reliable platform to mechanistically investigate the chemotherapeutic drugs on gastric cancer stem cells.
Export citation and abstract BibTeX RIS
Introduction
According to the National Institute of Health of the United States, gastric cancer remains a great clinical challenge because a surgical removal of stomach is the only means to completely cure the disease. Radiotherapy and chemotherapy can moderately contribute to the management of symptoms but are unlikely to cure the disease. Worse still, by the time the gastric cancer is diagnosed, it quite often has already metastasized to other organs, rendering the surgical removal of the stomach a meaningless treatment. Consequently scientists in both academia and the pharmaceutical industry have invested a paramount effort in discovering effective chemotherapeutic drugs or regimens for gastric cancer. To test the efficacy of interested drug molecules, a murine model is indispensable and critical because the readout determines whether or not the molecule warrants clinical trials down the road. An ideal murine model should possess a number of features for large-scale drug screening, such as a high incidence of tumor formation, a rapid tumor growth and a nice fidelity to human pathology to ensure a translational success. Unfortunately, however, current murine models used in cancer research fall short of these expectations, thus raising great concerns about their usefulness to churn out reliable chemotherapeutic drugs that could survive clinical trials [1–8]. The traditional xenograft tumor models comprise orthotopic and subcutaneous models. The orthotopic model is established by injecting tumor cells into the target organ whereas the subcutaneous model injects under the skin. Both of them enjoy many advantages, such as a rapid construction, the use of human tumor cells, etc. However, they both suffer from a wide range of limitations, such as a dysfunctional immune system, the lack of metastasis in the subcutaneous model, and surgical challenges in the orthotopic model, etc [1, 4, 7]. To overcome these hurdles, scientists tried to develop genetically engineered mice (GEM) as an alternative to the xenograft tumor models, arguing that GEM could recapitulate the entire cascade of pathological events in the tumorigenesis. However, after decades of research, the lack of data evidencing its superiority to the xenograft tumor models on predicting the efficacy of chemotherapeutic drugs in human patients has fueled a growing concern on its usefulness in cancer research. This disappointing performance can be largely attributed to the biological difference of tumors between human and mice [1, 2]. It does not help that the time and cost associated with breeding a sizeable colony of GEM for a large-scale drug screening is outright prohibitive for the pharmaceutical industry, further dampening its prospect in cancer research. Therefore, the xenograft subcutaneous model remains an indispensable and valuable platform for pre-clinical cancer research [9–11].
In this study, we aim to boost the tumorigenesis of the xenograft subcutaneous tumor model with an electrospun scaffold so that the effect of chemotherapeutic regimens can be accurately assessed. In the last two decades, tissue engineering scientists have strived to construct a great variety of scaffolds with proteins, polymers and inorganic materials (e.g. hydroxyapatite) to build a micro-environment analogous to the native extracellular matrix [12]. These tissue-engineered scaffolds have been employed to regenerate a wide range of lost or dysfunctional tissues, including bone, blood vessels, heart valves, etc [13–24]. Previous research has confirmed that tissue-engineered scaffolds could exert a profound influence on initiating and regulating the therapeutic tissue regeneration. For example, biochemical cues like growth factors can be incorporated into the scaffold and then regulate important cellular events, such as cancer metastasis, stem cell differentiation, etc [12, 25]. Based on findings from tissue engineering research, we hypothesized that the use of an electrospun scaffold would accelerate the tumorigenesis by facilitating the survival of implanted tumor cells and tested this hypothesis in both in vitro and in vivo systems.
Materials and methods
The electrospinning and physical characterizations of the scaffold
The scaffold was electrospun as previously described [20]. Briefly, polydioxanone (PDO) in the form of surgical sutures (Advanced Inventory Management, Mokena, IL), porcine gelatin A (Sigma-Aldrich, St. Louis, MO) and bovine elastin (Elastin Products Co. Inc., Owensville, MO) were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma-Aldrich, St. Louis, MO) to achieve a total concentration of 10% (w/v) (weight ratio = 8:1:2). The solution was loaded into a syringe capped with a 27-gauge needle mounted on a motorized pump (PHD 2000, Harvard Apparatus, Holliston, MA). A total of 1 mL of the blend solution was electrospun onto a grounded sheet collector at a feeding rate of 2 mL h−1 in a 1 kV cm−1 electric field. The electrospun scaffold was then desiccated in vacuum for at least 24 h. The morphology of the electrospun scaffold was studied with scanning electron microscope. The degradation of scaffolding materials was characterized by incubating the electrospun scaffold in tumor cell media at 37 °C and 5% CO2 for up to six weeks. At the end of each week, the sample was retrieved, desiccated in vacuum for 24 h and weighed on a digital balance. The weight loss was calculated as 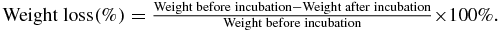
The collection of tumor cells and the sorting of CD24+ and CD44+ cells
Gastric tumors were resected from human patients at Shanghai Xinhua hospital. Tumor masses were immediately disassociated using sterile scissors and blades. To obtain a single-cell suspension, disassociated tissues were incubated with ultrapure collagenase IV (Worthington Biochemicals, Freehold, NJ) in medium 199 (200 units of collagenase per mL) at 37 °C for 4 h followed by a filtration through a 40 µm nylon mesh and a wash with HBSS/20% fetal bovine serum. The collected suspension was thoroughly rinsed with HBSS. CD24+ and CD44+ cells were sorted out by fluorescent activated cell sorting (FACS) (BD LSRII, BD Biosciences, San Jose, CA), using anti-human CD24 and anti-human CD44 antibodies (Pharmingen, Franklin Lakes, NJ). The sorting was repeated until the purification reached at least 90% in respective populations. Freshly sorted out CD24+ and CD44+ cells were used in the apoptosis assay in the chemotherapeutic effect study [26]. Written consent was obtained from patients prior to the resection of the pancreatic tumor specimens.
The viability and apoptosis analyses of gastric tumor cells on the scaffold
Desiccated scaffolds (D = 6 mm) were sterilized by in 70% ethanol followed by an extensive rinse in sterile phosphate buffered saline. The whole population of tumor cells were seeded on the scaffold (1000 cells/scaffold) and cultured for up to seven days for an in vitro tumorigenesis. 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (Promega, Madison, WI) and Caspase-Glo® 3/7 assay kit (Promega) were used to measure the viability and apoptosis, respectively, per manufacturer's protocols. Tumor cells cultured in tissue culture plates (TCP) were used as control.
The in vivo tumorigenesis of the gastric tumor in the scaffold model
Freshly resected gastric tumors from human patients were completely minced (∼1 mm × 1 mm) and cultured on sterilized scaffold (∼2 mm × 2 mm) at 37 °C and 5% CO2 overnight. Then the tumor/scaffold construct was surgically implanted subcutaneously into a BALB/c nu/nu female nude mouse (4–6 weeks old) (Shanghai Laboratory Animal Co., Shanghai, China). One edge of the scaffold was sutured to the skin to temporarily immobilize the construct. In the control group, each piece of the tumor tissue was implanted s.b. without the scaffold as in the traditional xenograft model. The animal study was approved by the Research Ethics and Compliance Committee at Shanghai Xinhua Hospital. The tumor site was photographed bi-weekly after the surgery and the tumor volume calculated as previously described: tumor volume = [L × W2]/2 [27]. By the end of week 12 post-surgery, all mice were euthanized and tumor masses retrieved and weighed on a digital scale prior to subsequent biological analyses.
Phenotypic characterizations of gastric tumor cells
The phenotype of cells in retrieved gastric tumor mass was determined by immunohistochemistry staining. Retrieved gastric tumors from mice were incubated in 10% formalin at 4 °C for at least 48 h, then embedded in paraffin and sliced into 4 µm thick serial sections on charged glass slides. For immunohistochemistry staining, sample were deparaffinized in xylene and rehydrated in ethanol gradients. Antigen retrieval was carried out by heating slides in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 15 min at 100 °C. Samples were blocked in 5% BSA buffer at 4 °C overnight. Thereafter, samples were incubated at 4 °C for 24 h with primary antibodies, including carbohydrate antigen 19–9 (CA199) (Abcam, Cambridge, MA), carcinoembryonic antigen (CEA) (Dako, Carpinteria, CA) and P53 (BD Pharmingen, San Jose, CA), which had been diluted in 1% BSA to recommended dilutions per manufacturers' protocols. At the end of the incubation, samples were extensively rinsed in PBS and then incubated at room temperature for 1 h with rabbit anti-human secondary antibodies conjugated with horseradish peroxidase (Abcam). All samples were counterstained with hematoxylin (Sigma Aldrich) before clearing and mounting. To measure the percentage of CD24+ and CD44+ cells, tumor masses were prepared as described above to obtain single-cell suspension and analyzed by flow cytometry using respective antibodies.
Chemotherapeutic effects of DCF and CF on gastric tumors in the scaffold model
Docetaxel, cisplatin and fluorouracil (DCF) and cisplatin and fluorouracil (CF) were both obtained from Shanghai Xinhua Hospital and diluted in sterile saline to indicated concentrations. The scaffold tumor model was established as described above. Implanted tumor cells were allowed to grow for four weeks before the drug administration. Immediately before the drug administration, the mice were randomly divided into three groups: DCF group (n = 7) that would receive docetaxel, cisplatin and fluorouracil, CF group (n = 6) that would receive cisplatin and fluorouracil, and NaCl group (n = 6) that would receive sodium chloride. In the DCF group, each mouse received by intravenous infusion docetaxel (2.14 mg kg−1) and cisplatin (2.14 mg kg−1) on day 1 followed by fluorouracil (21.4 mg kg−1 d−1) for three consecutive days. In the CF group, each mouse received by intravenous infusion cisplatin (2.86 mg kg−1) on day 1 followed by fluorouracil (28.6 mg kg−1 d−1) for three consecutive days. Dose of each drug was calculated based on previous research [28, 29]. The drug administration in both DCF and CF groups were repeated every two weeks for a total of four times by the end of the 8-week study. Mice in the NaCl group received equal volume of sodium chloride following the same drug administration schedule. All mice were euthanized after the 8-week-long drug administration. The tumor volume, weight, CD24+ and CD44+ cells were measured as described above.
In vitro anti-proliferation capacity of DCF and CF on CD24+ and CD44+ cells
CD24+ and CD44+ cells sorted out by FACS were seeded on sterilized scaffolds in a 96-well plate (5000 cells/well) and cultured for up to 48 h at 37 °C and 5% CO2. After seeding, samples were randomly divided into DCF, CF and NaCl groups (n = 3). In the DCF group, the regimen of docetaxel (200 µM), cisplatin (200 µM) and fluorouracil (2 mM) was added to the cell media at 8 h after seeding. Similarly, in the CF group, the regimen of cisplatin (300 µM) and fluorouracil (3 mM) was added at the same time point. In the control group, an equal volume of sterile NaCl was added to the cell media. The Caspase-Glo® 3/7 assay kit was used to measure the apoptosis immediately before the drug administration and after a 40 h drug treatment.
Data analysis
ImageJ was used to process and analyze all images. Student t-test and ANOVA with Tukey test were used where applicable (α = 0.05) in Prism (GraphPad Software, La Jolla, CA). All flow cytometric data was analyzed and graphically presented using FlowJo (Tree Star Inc., Ashland, OR). The median fluorescent intensity (MFI) of each population in each sample was measured by FlowJo and calculated using the formula below. An MFI% greater than 100% indicates the presence of interested cell population while an MFI% equal to 100% the lack of interested cell population. The MFI% increases along with the increase of the population size:
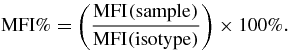
Results
The physical characterizations of the electrospun scaffold
The electrospun scaffold featured randomly distributed non-woven micro/nano-fibers with a highly porous structure, which mirrors the morphology of native extracellular matrix (figure 1(A)). It should be noted that each individual fiber was a composite of PDO, gelatin and elastin. We used PDO as the backbone of this electrospun scaffold to confer a structural stability to the scaffold while it would degrade in vivo without unleashing cytotoxic materials. To enhance cell adhesion and proliferation, we incorporated gelatin and elastin into the scaffold, two dominant proteins found in native extracellular matrix. This electrospun composite scaffold was extensively characterized by physical and chemical analyses in a previous research and showed promising capabilities to promote tissue regeneration [20]. Like most tissue-engineered scaffolds, we expect the scaffold to gradually degrade over the course of native tissue growth. Therefore, we measured the weight loss of this scaffold incubated in cell culture media to draw its degradation profile. The result showed that the weight loss reached 31.77% ± 7.29% within the first week and gradually climbed to 72.03% ± 12.45% by week 6, evidencing the degradability of scaffolding materials (figure 1(B)). The fact that the weight loss rate was high in the first week but much depressed thereafter could be attributed to the rapid disintegration of gelatin and elastin in the scaffold with a slow yet steady degradation of PDO. Scaffold samples became too fragile to manipulate for measurement after being incubated for over six weeks.
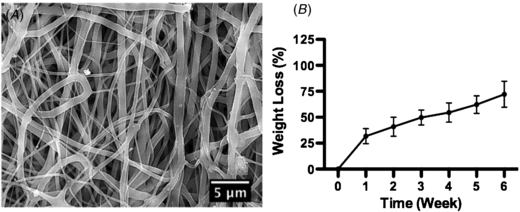
Figure 1. The physical characterizations of the PDO/gelatin/elastin scaffold. (A) SEM micrograph of the PDO/gelatin/elastin scaffold. (B) The weight loss in the in vitro degradation study. The micrograph showed that the scaffold was morphologically analogous to native extracellular matrix, featuring randomly distributed non-woven micro/nano-fibers. The degradation featured a higher rate in the first week due to the rapid disintegration of proteins. The degradation of the scaffold ensured an adequate time window for tumor cells to produce native extracellular matrix proteins. N = 3 in all groups.
Download figure:
Standard image High-resolution imageThe viability and apoptosis of gastric tumor cells on the scaffold
To support the tumorigenesis the scaffold must be able to support tumor cells of all types without producing a cytotoxic effect. To that end we cultured gastric tumor cells on the scaffold and measured the viability as well as apoptosis of the population. The population of tumor cells on the scaffold featured a higher viability than that on TCP on day 7 but no difference was observed on day 1, suggesting that the scaffold provided a more favorable environment than the TCP for cells to adhere and proliferate (figure 2(A)). A significant apoptosis was found in neither population on either day 1 or day 7, evidencing a lack of cytotoxic effects from scaffolding materials.
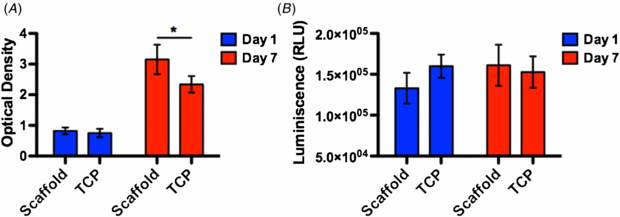
Figure 2. The viability and apoptosis analyses of tumor cells. (A) The viability analysis. (B) The apoptosis analysis. The viability analysis showed that tumor cells on the scaffold outgrew their counterparts on TCP on day 7, suggesting that the scaffold was a more favorable substrate for cells to grow. No significant apoptosis was detected in either group on either day, evidencing a lack of cytotoxic effect of scaffolding materials. A star sign indicates a statistical difference between groups by Student t-test (α = 0.05). N = 3 in all groups.
Download figure:
Standard image High-resolution imageIn vivo tumorigenesis in the scaffold model
The in vivo study demonstrated that this electrospun composite scaffold enhanced tumorigenesis through week 12 post-surgery (figures 3(A) and (B)). The tumor volume in the scaffold group started to out-grow its peers in the traditional tumor group on week 8 post-surgery and sustained this advantage until week 12. By week 12 after the surgery, the tumor volume and weight in the scaffold model group was 43.27% and 75.58% greater than that the traditional model group, respectively. This increased tumorigenesis in the scaffold model substantiated our hypothesis that the use of an electrospun scaffold would facilitate the tumor growth.

Figure 3. The in vivo tumor growth of implanted tumor cells. (A) The tumor volumes in the scaffold/tumor model (n = 8) and traditional tumor model (n = 7). (B) The tumor weight on week 12. Digital photographs of gastric tumor on week 12 post-surgery from the scaffold/tumor group (C) and the traditional tumor group (D). From week 8 on, the gastric tumor in the scaffold/tumor group was consistently larger than that in the tumor group. By week 12, the tumor volume in the scaffold/tumor group was 143.27% of that in the tumor group. The tumor weight from the scaffold/tumor group was 75.58% greater than that from the tumor group. These results showed that the scaffold promoted the gastric tumorigenesis. A star sign indicates a statistical difference between groups by Student t-test (α = 0.05).
Download figure:
Standard image High-resolution imageThe phenotype of cells in the gastric tumor mass
Previous research demonstrated that instructive information conveyed from tissue-engineered scaffolds could dictate cellular events like stem cell differentiation [12]. To detect if scaffolding materials led to unwanted phenotypic change, we probed the expression of gastric tumor markers, CA199, CEA and P53 (figure 4). All three markers were highly expressed in cells in the gastric tumor tissues in the scaffold model group as those the traditional model group on week 12 post-surgery. This result evidenced that neither the chemistry of scaffolding materials nor the physics of the scaffold caused aberrant phenotypic changes during the tumorigenesis, thus offering cancer scientists a reliable platform to evaluate chemotherapeutic drugs. The enhanced expression of P53 in the scaffold group can be attributed to the use of the scaffold, a variance among individuals or both.

Figure 4. Immunohistochemistry staining of CA-199, CEA and P53 in gastric tumor cells on week 12 post-surgery. CA-199 (A) and (B), CEA (C) and (D) and P53 (E) and (F). Samples (A), (C) and (E) were from the scaffold/tumor group; samples (B), (D) and (F) were from the tumor group. The significant expression of CA-199, CEA and P53 in the scaffold/tumor group confirmed that no aberrant phenotypical changes occurred due to the scaffold.
Download figure:
Standard image High-resolution imageThe characterization of CD24+ and CD44+ cells in the tumor mass
In gastric cancer, CD24+ and CD44+ cells in the tumor mass were believed to the most potent cancer stem cells (CSC) that drives the tumorigenesis in vivo [30, 31]. Therefore, we investigated the influence from the scaffold on the proliferation of CD24+ and CD44+ cells. The flow cytometric analysis showed that the scaffold model saw an increased percentage of both CD24+ and CD44+ populations by the end of the 12-week study (figure 5). The histogram of CD24+ and CD44+ cells in the scaffold model showed a wider shift than those in the traditional model group, indicating a larger population in the scaffold model. A quantitative analysis of the MFI, a widely used benchmark in flow cytometric analysis, showed that CD24+ and CD44+ populations in the scaffold model saw a 42% and 313% increase, respectively, compared to those in the traditional model. This increased proliferation of CSC in the tumor in the scaffold model could have greatly contributed to the accelerated tumorigenesis.

Figure 5. The populations of CD24+ and CD44+ CSC in the tumor . (A) Representative flow cytometric histogram of CD24+ and CD44+ populations in gastric tumors retrieved on week 12 post-surgery. Blue: sample; red: isotype control. The degree of shift of blue from red corresponds to the size of respective population. (B) The statistical analysis of MFI of CD24+ and CD44+ populations. The MFI of the CD24+ population in the scaffold/tumor group (n = 8) was 42% higher than that in the tumor group (n = 7). More strikingly, the CD44+ population saw a 313% increase of MFI when the scaffold was employed. This MFI increase evidenced that the population of CD24+ and CD44+ in the scaffold/tumor model witnessed a greater proliferation. A star sign indicates a statistical difference between groups by Student t-test (α = 0.05).
Download figure:
Standard image High-resolution imageThe evaluation of chemotherapeutic effects of DCF and CF on gastric tumor
After confirming the reliability and superiority of our scaffold model, we took advantage of it to comparatively investigate chemotherapeutic effects of DCF and CF regimens on gastric tumors. Both DCF and CF unequivocally arrested the growth of gastric tumor while the tumor underwent a steady and fast growth in the NaCl group (figure 6(A)). A significant difference between chemotherapeutic groups and NaCl group emerged at week 6 and was further pronounced by week 8. At the end of the 8-week treatment, tumor volumes in the DCF and CF groups were only 21.88% and 28.58% by average of that in the NaCl group, respectively. Correspondingly, tumor weights in DCF and CF groups were 26.03% and 32.25% of that from the NaCl group (figure 6(B)). The traditional tumor model showed a delayed onset of the chemotherapeutic effect of both DCF and CF as evidenced by the emergence of tumor volume difference as late as week 8 and a large variance within treatment groups, indicating a poor reliability (figure S1 (available from stacks.iop.org/BMM/8/045003/mmedia)). These results confirmed that both DCF and CF unleashed a proven chemotherapeutic effect on gastric tumor to arrest its growth and that the scaffold model was a robust and reliable platform for drug studies. These results prompted us to hypothesize that the arrest of tumor growth could be attributed to the chemotherapeutic effect on CD24+ and CD44+ CSC in the tumor masses. The flow cytometric analysis confirmed our hypothesis. Both DCF and CF diminished the CD24+ and CD44+ CSC populations after an 8-week treatment (figure 7(A)). The MFI analysis revealed that the DCF possessed a stronger ability than CF to deplete the CD44+ population whereas both showed similar competency in depleting CD24+ cells (figure 7(B)). To verify this finding, we measured the apoptosis level of CD24+ and CD44+ CSC due to DCF and CF treatment. The measurement agreed with the in vivo finding by showing that DCF induced a greater apoptosis of CD44+ cells than CF when no difference was observed prior to the drug treatment (figures 7(C) and (D)). However, the apoptosis levels of CD24+ cells were similar between DCF and CF treatments.
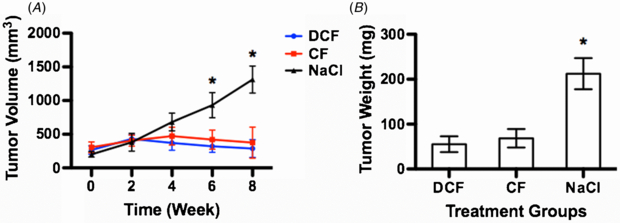
Figure 6. The chemotherapeutic effect of DCF and CF on tumor growth in the scaffold model. (A) The tumor volume during an 8-week treatment. (B) The tumor weight on week 8. The tumor volumes of DCF and CF treated mice were 21.88% and 28.58% of those of NaCl treated mice, respectively, by week 8. The tumor weights in DCF and CF treated mice were 26.03% and 32.25% of that in NaCl treated mice, respectively, on week 8. These results showed that both DCF and CF effectively capped the growth of tumor. A star sign indicates a statistical difference among groups determined by ANOVA test followed by Tukey test (α = 0.05). DCF group (n = 7); CF group (n = 6); NaCl group (n = 8).
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. The chemotherapeutic effect of DCF and CF on CD24+ and CD44+ cells. (A) Representative flow cytometric histogram of CD24+ and CD44+ populations in each treatment group. Blue: sample; red: isotype control. (B) The statistical analysis of MFI of CD24+ and CD44+ populations on week 8. DCF virtually depleted both CD24+ and CD44+ cells by week 8 whereas CF only moderately reduced CD44+ cells despite its depleting effect on CD24+ cells. The MFI of CD44+ cells in the DCF and CF groups were 17.93% and 30.97% of that observed in the NaCl group; the MFI of CD24+ cells in the DCF and CF groups was 40.27% and 59.94% of that in the NaCl group. (C) The apoptosis level of CD24+ and CD44+ cells before drug administration. (D) The apoptosis level of CD24+ and CD44+ cells after drug administration. No significant apoptosis in any group was observed before the drug administration. However, apoptosis spiked in DCF and CF treated CD24+ and CD44+ cells. Particularly, DCF induced a greater apoptosis than CF in CD44+ cells. In the in vivo study, DCF group (n = 7); CF group (n = 6); NaCl group (n = 8). In the apoptosis study, n = 3. A star sign indicates a statistical difference between groups by ANOVA test followed by Tukey test (α = 0.05).
Download figure:
Standard image High-resolution image{kind=link}
Discussion
In tissue engineering research, a scaffold is expected to provide a temporal physical environment for cells to adhere, migrate and proliferate, and should degrade to accommodate the production of native extracellular matrix proteins [32]. In this study we selected PDO, gelatin and elastin as the components of the electrospun scaffold based on previous research [20]. The PDO/gelatin/elastin scaffold featured a highly porous microstructure analogous to the native extracellular matrix, which is expected to promote cell adhesion and migration (figure 1(A)). PDO is a degradable synthetic polymer widely used in surgical sutures for its excellent mechanics and biocompatibility for in vivo applications. To our knowledge, no cytotoxic effects from PDO have been reported, making it an ideal candidate for scaffolding materials. Our previous research found that proteins in the scaffold would disintegrate roughly within a week after being introduced into an aqueous media [21]. Consequently, the presence of PDO in the scaffold preempted a rapid structural disintegration of the scaffold, thus providing cells a sufficient time window to produce their native extracellular matrix proteins (figure 1(B)). However, PDO lacks biological domains that are crucial to facilitate cell-scaffold interactions, such as adhesion, migration, etc. To fill this gap, we incorporated gelatin and elastin, two primary proteins in native extracellular matrix, into the scaffold to present active motifs to facilitate cell activities. Our results showed that tumor cells on the scaffold outgrew their counterparts on TCP within a week suggested that these proteins had contributed to the adhesion and then survival of tumor cells, which ultimately translated into an increased proliferation (figure 2). The in vivo study mirrored the findings from in vitro studies that the scaffold could promote tumor growth. Tumors implanted with scaffolds outgrew those in the traditional tumor model, evidencing the scaffold accelerated the tumorigenesis (figure 3). Also, the scaffold-supported gastric tumor was phenotypically identical to its peer in the traditional model (figure 4). Challenges associated with the traditional tumor model are that the incidence of tumor is unpredictable and that the implanted tumor takes months to grow to an adequate size for chemotherapeutic studies. We attribute the low reliability and slow tumorigenesis to the unfavorable subcutaneous space for delivered cells to rapidly adhere and proliferate. The scaffold afforded a favorable microenvironment for cells in the initial stage after implantation, effectively led to an increased survival and proliferation of tumor cells.
In recent years, cancer scientists discovered that the tumor is made of a large heterogeneous population of cells and argued that a sub-population of those cells, CSC, is the driving force for tumorigenesis [33–35]. With the research on CSC exploding, the nature and functions of CSC have led to a hot debate. Now the consensus tells that the CSC can be broadly defined as the sub-population of cells in the tumor that possess a tremendous capacity of self-renewal, multi-lineage differentiation, and regulation of genes involved in the self-renewal of normal stem cell [33, 34]. Unfortunately, the research on associating certain surface markers to CSC and the roles of different subsets of CSC in tumorigenesis is far from being conclusive. Despite many surface markers are commonly expressed in CSC, the latest wisdom goes that each type of cancer features its own combination of surface markers, for example, gastric CSC readily expresses CD24 and CD44 [30, 31]. Moreover, CSC are not born equal, for example, CXCR4+ CSC primarily drives metastasis [36, 37]. Even though the perspective into CSC remains limited, the very existence of CSC and their roles in tumorigenesis has been widely believed to be a promising therapeutic target to defeat cancer. Illuminated by previous research, we set forth to explore the underlying mechanism of the accelerated tumorigenesis in our scaffold model, focusing on CD24+ and CD44+ cells. We investigated CD24+ and CD44+ cells, rather than narrowly focusing on CD24+CD44+ cells, based on the fact that both populations possessed the nature of CSC but might respond to the same chemotherapeutic regimens differently. In addition, the percentage of CD24+ cells ranged from 5% to 23% in the tumor whereas CD44+ cells from 3% to 11%. In contrast, CD24+CD44+ cells were quite rare, ranging from 0.2% to 2.7%. Consequently, the exploration of how to deplete a sizeable population of CSC (CD24+ and/or CD44+ cells) bears a much greater clinical significance than only targeting the tiny population of CD24+CD44+ cells. The flow cytometric analysis showed that both CD24+ and CD44+ populations saw a significant increase in the scaffold model with the increase of CD44+ cells particularly pronounced (figure 5). This increase of CSC could have contributed to the accelerated tumorigenesis observed in the scaffold model.
Thereafter, we proceeded to comparatively investigate the chemotherapeutic effect of DCF and CF. Both regimens have been studied for advanced gastric cancer and showed clinical effectiveness to some extent [28, 29, 38, 39]. However, little is known about the respective underlying mechanism, which is critical to further modify the formulation to improve the clinical performance and to minimize toxic side effects. Our study found that DCF and CF both capped the growth of gastric tumors, which could be attributed to their ability to deplete CD24+ and CD44+ cells (figures 6 and 7). It is of particular interest that DCF unleashed a greater power to eliminate CD44+ cells than CF, which could be accounted for by DCF's ability to induce a greater apoptosis in CD44+ cells. While the presence of docetaxel in the DCF regimen boosted its chemotherapeutic effect on CD44+ cells, it should be noted that this improvement could be attributed to docetaxel as well as its interactions with cisplatin and fluorouracil. This finding will facilitate the modification of existing regimen and the development of novel regimen based on cisplatin and fluorouracil to maximize the chemotherapeutic effect on CSC while minimizing toxic effects.
In this study, we revamped the traditional tumor model using an electrospun scaffold and saw an accelerated tumorigenesis in vivo. This scaffold model has proven to be a reliable and powerful platform to investigate chemotherapeutic regimens. However, we are also keenly aware of limitations of the xenograft subcutaneous model. For example, the immune system of the host mice is dysfunctional to allow the growth of implanted human tumor. The lack of a functional immune system generates a ripple effect on how cancer cells are dealt with by the host. Genetically induced tumors in GEM with a competent immune system are biologically different from human tumor, irking many cancer scientists. Therefore, xenograft remains an indispensable tool in cancer research and a popular one for the pharmaceutical industry for economic reasons. Our revamped model thus could contribute to the cancer research by producing reliable results of drug screening and in-depth perspectives into chemotherapy.
Acknowledgments
The authors declare no conflict of interests. The authors acknowledge Dr Xing Zhang at the University of Chicago for his valuable suggestions on the study and the manuscript preparation. The authors acknowledge the financial support from China's National Natural Science Foundation (grant numbers: 30300357 and 30801107) and the Ministry of Education of Shanghai Municipal Government (grant number: 09YZ84).